Decode of trajectory
Posted: Mon Jan 20, 2014 4:04 am
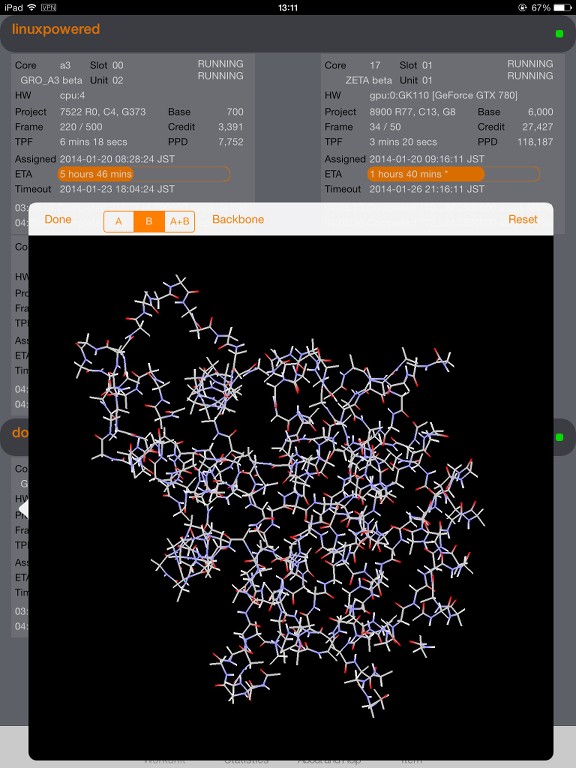
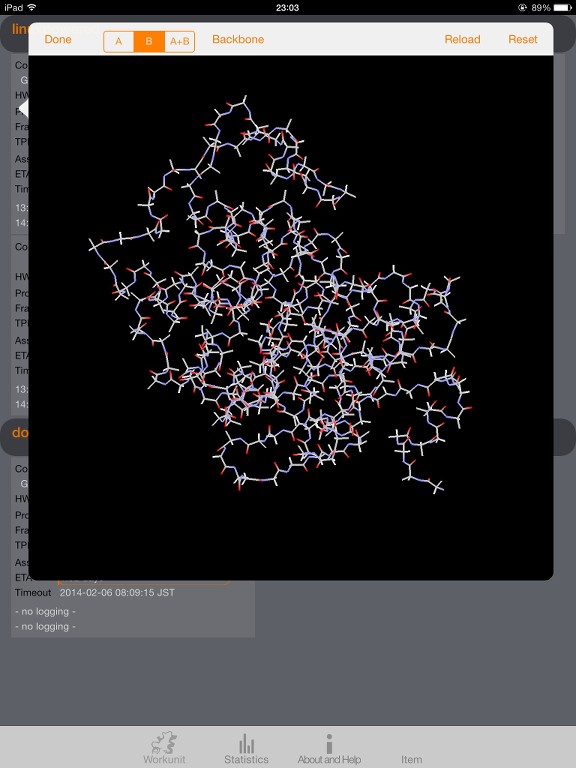
I start to understand the data provided by the trajectory on high level and use it for visualization; right now in a more simple way (atoms with and without bonds).
I wonder if there is an algorithm described and available how to decode the atoms and bonds and figure out where is the backbone(s) of the protein and what side chains are connected.
I have some draft algorithm for the backbone but it's too rough as it's mainly focus on atom numbers and surrounding atoms, looking for N and C as potential backbone candidates. Not very precise as I can see small orphans in my model. Some side chains, when locally checked with my beginner algorithm, appears as backbone.
I'm sure there is something better around ... Any hint would be welcome
I wonder if there is an algorithm described and available how to decode the atoms and bonds and figure out where is the backbone(s) of the protein and what side chains are connected.
I have some draft algorithm for the backbone but it's too rough as it's mainly focus on atom numbers and surrounding atoms, looking for N and C as potential backbone candidates. Not very precise as I can see small orphans in my model. Some side chains, when locally checked with my beginner algorithm, appears as backbone.
I'm sure there is something better around ... Any hint would be welcome